IRB Member Resources
UC Davis IRB Members have superior knowledge, experience and dedication. We appreciate their hard work and thorough review of proposed research. The following are useful resources for conducting IRB reviews.
In This Section
- Reviewer’s Guide
- Choosing Determinations
- Definition of Minimal Risk
- Reviewer’s Checklists
- Committee Member Tip Sheet
- Review Guidance for Special Topics
Reviewer’s Guide
Initial Submissions
Renewal Submissions
Modification
Reportable New Information
Consent Template
Worksheets
- HRP-306 Drugs
- HRP-307 Devices
- HRP-314 Criteria for Approval and Additional Considerations
- HRP-314B Requirement for Informed Consent
- HRP-315 Advertisements
- HRP-316 Payments
- HRP-318 Additional Federal Agency Criteria
- HRP-325 Clinical Trials Contraception
Choosing Determinations
Approval or Approval with Administrative Comments
An “approval” or “approval with administrative comments” is issued when the committee has no questions or concerns about the current submission. The IRB members may provide comments about the approval or recommendations for future submissions. These comments will be included in the approval letter.
Modifications Required to Secure Approval
Granted when the submission does not meet the criteria for approval, Worksheet: Criteria for Approval (HRP-314), but committee recommends approval contingent on required directed revisions to the protocol or consent document. When this determination is made, the response does not need to go back to Committee for review. The required revisions will be described in the letter of action. The investigator must provide a separate, point-by-point response. The revisions must be made exactly as described in the letter of action in order for the study to be approved by a designated reviewer without additional review by the committee. If the revisions submitted in the response are not exactly as described in the letter of action, or if the response contains revisions in addition to those requested, the response will be assigned to an agenda for full committee review.
Deferral
Determination for research that does not meet the criteria for approval, Worksheet: Criteria for Approval (HRP-314), and new information or clarification is needed to determine that one or more of the criteria are satisfied. This determination is made when serious concerns are raised about the risk/benefit ratio or other issues of human subject protection and the members of the committee agree that additional information, justification, or changes are needed before approval can be reconsidered. The investigator’s response to the committee’s requirements will be reviewed by the full committee.
Disapproval
The committee has found that the study design, research risks and benefits, consent process, and/or documentation of consent are inappropriate or inadequately described, such that the committee cannot specify changes necessary to meet the criteria for approval, Worksheet: Criteria for Approval (HRP-314). This determination is made very rarely. The investigator’s response to the committee will typically require major revision to the study’s procedures, inclusion/exclusion criteria, or aims and will need to be reviewed by the full committee.
Non-Compliance
- A failure to comply with the requirements of an applicable law, regulation, or institutional policy pertaining to the protection of human subjects, or with the requirements or determinations of an IRB, that poses a risk of harm to subject’s rights, safety or welfare, or the integrity of the research data. Non-compliance may be the result of the action or inaction of anyone conducting protocol procedures, but not research subjects.
- In the case of research funded or conducted by the Department of Defense (DOD), Non- Compliance includes failure of a person, group, or institution to act in accordance with Department of Defense (DOD) instruction 3216.02, its references, or applicable requirements.
Serious Non-Compliance
Noncompliance that adversely affects the rights or welfare of participants. For Department of Defense (DOD) research Serious Non-Compliance includes failure of a person, group, or institution to act in accordance with Department of Defense (DOD) Instruction 3216.02 and its references such that the failure could adversely affect the rights, safety, or welfare of a human subject; place a human subject at increased risk of harm; cause harm to a human subject; affect a human subject’s willingness to participate in research; or damage or compromise the scientific integrity of research data.
Continuing Non-Compliance
A pattern of noncompliance that indicates an inability or unwillingness to comply with the requirements of an applicable law, regulation, or institutional policy pertaining to the protection of human subjects and/or with the requirements or determinations of an IRB.
Unanticipated Problem Involving Risks to Subjects or Others
An incident, experience, or outcome that meets all of the following criteria:
- Unexpected (in terms of nature, severity or frequency) given (a) the research procedures that are described in the protocol-related documents, such as the IRB-approved research protocol and informed consent document; and (b) the characteristics of the subject population being studied;
- Related or possibly related to participation in the research (possibly related means there is a reasonable possibility that the incident, experience, or outcome may have been caused by the procedures involved in the research); and
- Suggests that the research places subjects or others at a greater risk of harm (including physical, psychological, economic, or social harm) than was previously known or recognized, even if no harm has actually occurred.
None of the Above (Acknowledgement)
None of the Above (Acknowledgement)
Definition of Minimal Risk
The probability and magnitude of harm or discomfort anticipated in the research that are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.
Reviewer’s Checklists
Federal regulations require the IRB to make specific determinations for waivers of consent or documentation of consent, for inclusion of certain vulnerable populations and for investigations of drugs and devices. UC Davis IRB uses a checklist to document these determinations in a manner acceptable to regulators. Members are asked to complete the following documents, when applicable.
- HRP- 410 Waiver or Alteration of the Consent Process
- For use when consent will be waived or when deception is used.
- HRP- 411 Waiver of Written Documentation of Consent
- For use when the requirement for a signature on a consent form is waived.
- HRP-412 Pregnant Women
- HRP-416 Children
- For use when individuals who meet the definition of “child” will be included in the research.
- Children means persons who have not attained the legal age for consent to treatments or procedures involved in clinical investigations, under the applicable law of the jurisdiction in which the clinical investigation will be conducted.
- HRP-417 Cognitively Impaired Adults
- For use when individuals who lack capacity to personally consent are included in the research.
- HRP-418 Non-Significant Risk Device
- For use with clinical investigations of medical devices
- HRP-441 HIPAA Waiver of Authorization
- For use when identifiable medical records will be accessed without authorization
Committee Member Tip Sheet
Review Guidance for Special Topics
Contraceptive Use
Appropriate contraceptive measures must be practiced when participation in a study could adversely affect an embryo, fetus, breastfeeding baby, or if it is unknown whether such harm could occur. Below is general guidance regarding how the IRB will evaluate risks regarding contraception use.
Contraceptive Options
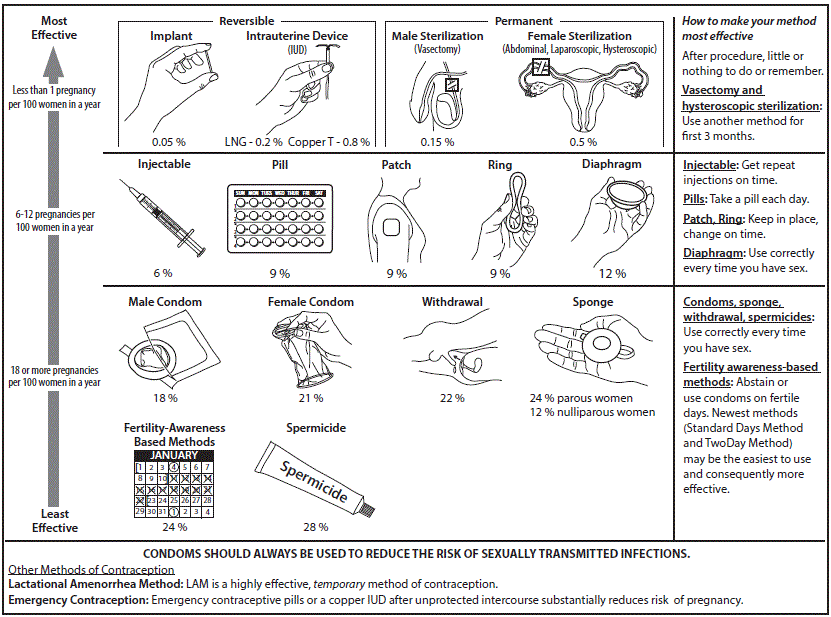
If a subject is able to become pregnant and must avoid becoming pregnant while on a study, the IRB will usually require at least one of the following methods of contraception:
Highly Effective
- Vasectomy
- Contraceptive implant
- Implantable hormone
- Intrauterine device “IUD” with hormones
- Intrauterine device “IUD” without hormones
Moderately Effective
- Hormonal injection
- Oral contraceptives, “the pill”
- Vaginal ring
- Contraceptive patch
In most instances, a protocol will require “highly effective contraception” as defined by the CDC. However, if the contraception options include methods other than highly effective methods the IRB will determine if the listed options are acceptable or NOT acceptable.
Contraindicated Methods and Increased Risk for Being in the Study
In some instances, certain birth control methods may be contraindicated for the subject population included in the study. For example, contraceptives containing estrogen may be contraindicated in studies of persons with cancer. Researchers should consider such contraindications when writing a protocol.
Acceptable methods and contraception requirements for studies are considered par of the eligibility criteria of a study.
Provisions for Pregnancy Testing
If a study requires subjects to avoid causing a pregnancy or becoming pregnant, the protocol should include adequate provisions for pregnancy testing. A pregnancy test should be performed before a subject who is able to get pregnant begins study procedures. Pregnancy tests may also be required throughout participation in the study.
Considerations for the Informed Consent Document
If a study has a contraception requirement, the consent form must include information about the contraception requirement for subjects able to become pregnant and contraception requirements for subjects able to cause a pregnancy.
Sponsors typically provide language about contraception requirements in the sponsor’s version of the consent form. However, in circumstances when such language is not available you may use the IRB template language found in Consent Form Sample Risk Language.
Please note, if the protocol or Investigator’s Brochure requires two methods of birth control, then the UC Davis IRB permits two options for subjects:
- Use of one method of highly effective contraception, or
- Use of one moderately effective method of contraception in conjunction with a barrier method (e.g., condom)
Contraception Information Sheet
The IRB requires researchers complete and submit a Contraception Information Sheet with each initial submission when the following criteria is met:
- Any study with contraception requirements for partners who are able to become pregnant or,
- In which a condom is required to protect from semen exposure
Subjects who are able to cause a pregnancy should receive an Information Sheet from the research team describing the contraception requirements to provide to their partner(s) who are able to become pregnant.
Pregnant Partner Consent Forms
The pregnant partner consent forms should only be submitted to the IRB in an instance when a partner becomes pregnant while the subject is on study or during the timeframe restricted by the study. The IRB strongly encourages research teams to refrain from submitting a pregnant partner consent form at initial review. If the Sponsor requires IRB review and approval, the IRB will still review the document. However, submitting the pregnant partner consent form could delay approval of the initial submission.
Gender Neutral Language
Gender neutral language is only required in consent forms for initial submissions. It is not required on studies with consent forms that have been previously approved.
Reviewer Resources
Worksheet HRP-325 can also help reviewers when conducting their review and considerations on whether risks are minimized.
Skin Biopsy Risk Determination
The UC Davis IRB strives for consistency when evaluating the risks of research procedures. The risk level of all procedures are evaluated by taking into account the procedure, subject population, setting, and personnel performing the procedure. Below is general guidance regarding how the IRB will evaluate risks of skin biopsies. Study-specific determinations may vary.
Some subjects may be prone to excessive scar formation. When there is no direct benefit from participation, potential subjects with these conditions should not be enrolled in studies that require skin biopsy. Inclusion/exclusion criteria for the study should note that subject with these conditions will be excluded. Additionally, underlying conditions of the subject may make the procedure greater than minimal risk (e.g. coagulopathy, hemophilia). If these underlying conditions are a focus of the study, these conditions and possible complications should be taken into account when making a risk determination.
Minimal Risk
Skin biopsies can be determined to be a minimal risk procedure if:
- The skin biopsies are required to meet the aims of the research.
- There is no less traumatic alternative biological sample (such as blood) that could serve the same purpose.
- The biopsies are 2 mm in diameter or less and require not sutures.
- Appropriate anesthetic will be applied before the procedure.
Greater than Minimal Risk
Skin biopsies not meeting the four criteria above should be considered greater than minimal risk. Additionally, if the study targets a condition which makes the acquisition of even small biopsies without a suture problematic, this should be taken into account.
Children Risk Level Determinations
Any skin biopsy that requires suturing in a study that does not have the prospect of direct benefit to the child participants will be considered a minor increase over minimal risk.
Considerations for the Informed Consent Document
- The size of the biopsy should be explained in a measurement that is understandable to the population. For example, “The size of the biopsy that will be taken is 0.08 inches in diameter.”
- The consent form should specify that obtaining skin biopsies may cause some discomfort similar to a blood draw, however it is important to note that subjects will need to keep the area clean and cover the site with a bandage for a longer period of time than a blood draw.
See “Skin Biopsy Risks” in Consent Form Sample Risk Language for example language.
Electronic Devices
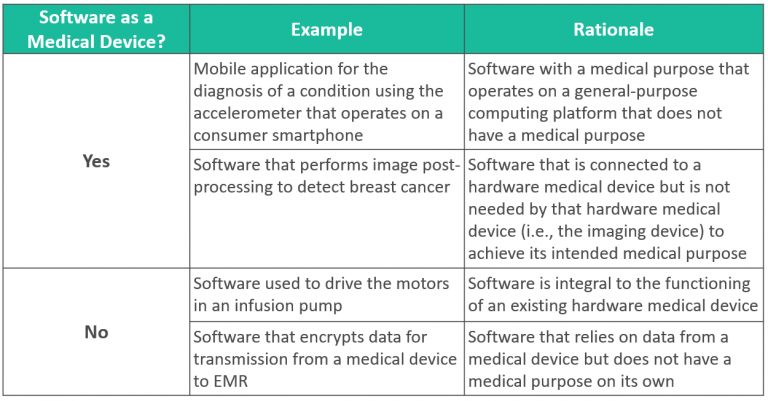
Software as a Medical Device
Software, including mobile medical applications, may be classified as medical devices. Software is NOT a medical device if:
- Software is integral to the function of a hardware medical device, and/or
- Software does not have a medical purpose on its own.
Considerations for the Informed Consent Document
Any other terms and conditions the subject must agree to in order to use the device should be mentioned in the informed consent document. These other terms and conditions do not need to be evaluated by the UC Davis IRB unless they are specific to the research. For example, the terms and conditions for the use of a particular wearable tracker do not need to be reviewed by the IRB if those terms and conditions are signed by all users, regardless of their participation in the research.
Information Technology Evaluation
IT evaluation is required when research includes implementation or use of new applications, systems or devices that collect and/or transmit sensitive identifiable information such as Protected Health Information (PHI) or Personally Identifiable Information (PII).
If the electronic device will use or record identifiable information, then must undergo information technology evaluation.