Full Committee Review Information
The IRB has two full board committees that review all human subject research studies that cannot be reviewed under the expedited review process. The guidelines below describe how our office assigns protocol submissions for review. Please note that these are guidelines and can be adjusted based on concern for subject safety, requests for rush review, and submission volumes.
In This Section
- How are Studies Assigned to a Full Committee Agenda?
- What Types of Studies are Reviewed at a Full Committee Meeting?
- How Reviewers Evaluate a Study
- Expiration Dates
- IRB Fees
- Becoming an IRB Member
- Special Situations
Related Topics
How are Studies Assigned to a Full Committee Agenda?
Committee agendas are scheduled and sent out to reviewers about 7-10 days prior to the meeting date. Submissions will be assigned to an agenda based on this timeframe.
Biomedical/Clinical Research
Biomedical research is the study of specific diseases and conditions, both mental and physical. This kind of research can encompass the detection, cause, prophylaxis, treatment and rehabilitation; the design of methods, drugs and devices used to diagnose, support and maintain the individual during and after treatment for specific diseases or conditions; and the scientific investigation used to understand the underlying processes which affect diseases, including the cellular and molecular bases of diseases, genetics and immunology.
Social & Behavioral Research
Social & Behavioral research refers to research that deals with human attitudes, beliefs, and behaviors. This kind of research is often characterized by data collection methods such as questionnaires, interviews, focus groups, direct or participant observation, and non-invasive physical measurements. The research may be qualitative or quantitative and may also include epidemiological, outcomes, or health services research.
Research on Human Nutrition and Supplements
Research on Human Nutrition and Supplements usually examines the effects of food, diet, and nutrition on human health. These studies often involve dietary supplements, which are regulated differently than drugs. While drugs are intended to diagnose, cure, mitigate or treat, or prevent any disease, a supplement can affect the structure and function of the body but cannot have a therapeutic purpose. The intended use of the product, as well as any labelling and advertising will ultimately determine whether or not it is considered a drug. Please contact the IRB for guidance on whether or not your study is exempt from the FDA’s drug regulations.
What Types of Studies are Reviewed at a Full Committee Meeting?
Why do Studies Get Reviewed by the Full Board?
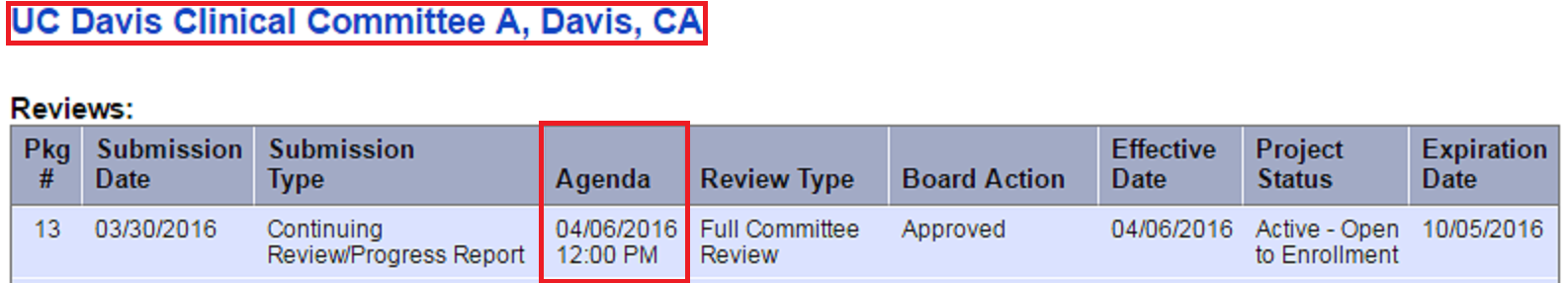
Both of our IRB Committees review studies that are or may involve more than minimal risk to subjects. If a modification has the potential to increase the risk/benefit ratio, it will be referred to the Full Committee by an expedited reviewer (Worksheet: Eligibility for Review Using the Expedited Procedure (HRP-313)). Once a package has been assigned to a Full Committee agenda, the reviewing board and agenda can be located within IRBNet within the “Review” section. Instructions as follows:
- Enter your user name and password at irbnet.org
- Click the study title to open the protocol
- Click “Reviews”, within the left side toolbar
- Look for the submitted package
How Do Studies Get Reviewed by the Full Board?
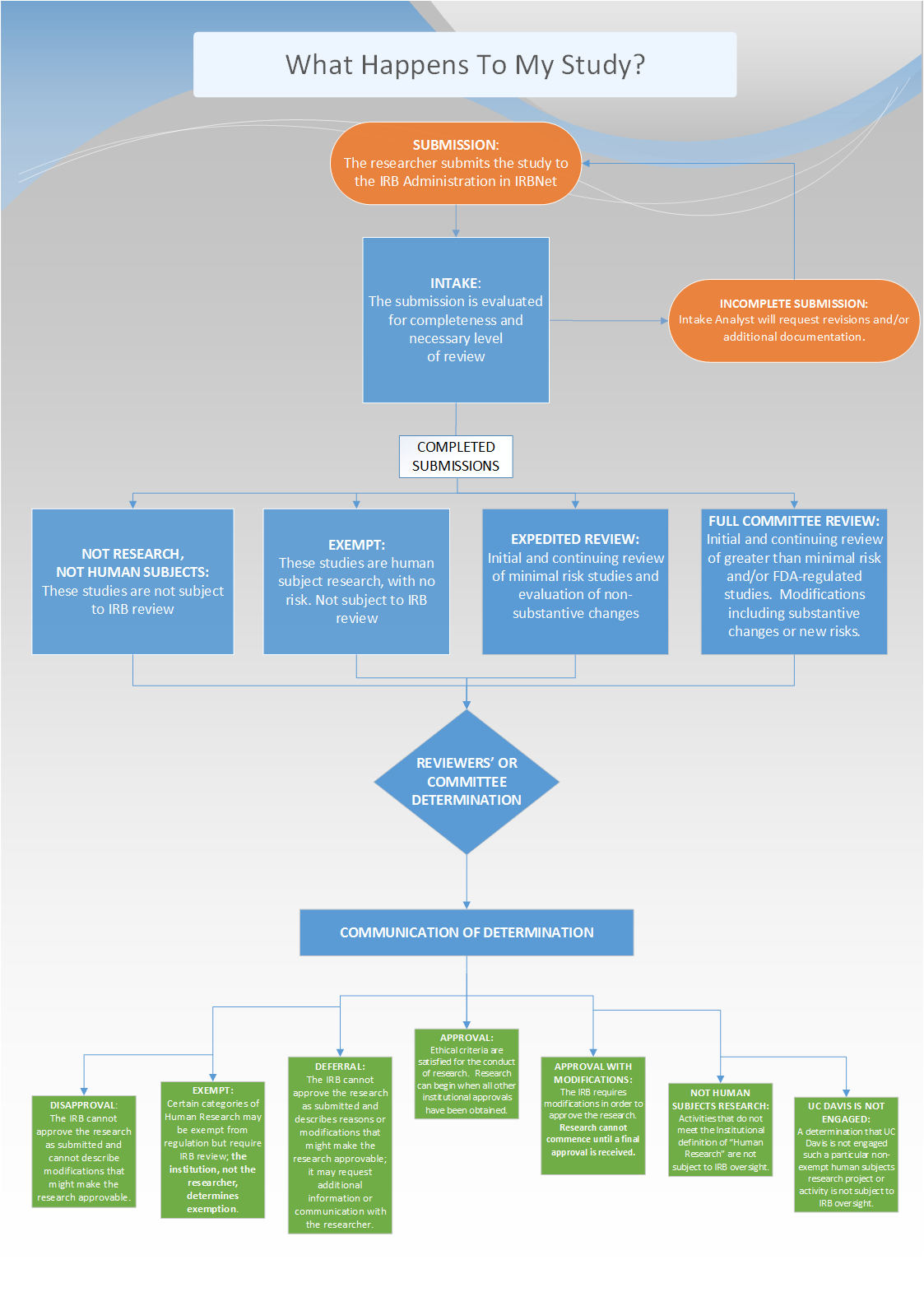
View the flow chart in PDF form here
Initial Applications (New Projects)
Assigned to a committee based on subject, the date received, and whether the appropriate expertise is scheduled to attend. Approximately 6 initial submissions will be scheduled per meeting, although this may change, depending on volume.
Modifications/Amendments
Assigned to the next available agenda for the committee that reviewed the most recent approved submission. These items may occasionally be assigned to an agenda on short notice, or assigned to a different clinical committee, depending on the severity of issues being reported or the timeline required for the implementation of modifications.
Previously Reviewed and Returned to Committee
Responses to a previous determination of Deferral or when the investigator chooses not to make the changes required for contingent approval. Assigned to the next available agenda for the committee that reviewed the previous submission.
Continuing Reviews/Study Closures
Greater than minimal risk studies usually require annual review by the full committee, with four exceptions:
- No subjects have been enrolled at UC Davis and no additional risks have been identified from any site or other relevant source.
- The research is permanently closed to the enrollment of new subjects, all subjects have completed all research-related interventions, and the research remains active only for long-term follow-up of subjects.
- The research is permanently closed to the enrollment of new subjects and the remaining research activities are limited to data analysis.
Studies will be assigned to an agenda based on expiration date and volume of submissions, but usually about 4-6 weeks prior to expiration. Continuing review submissions containing substantive modifications or new risks will be prioritized for Full Committee review.
Reportable New Information
Assigned to the next available agenda for the committee that reviewed the previous submission. These items may be assigned to an agenda on short notice, or assigned to a different clinical committee, depending on the severity of issues being reported.
How Reviewers Evaluate a Study
Research studies are submitted for full board review following screening by IRB Administration staff and review by Committee Analysts. Studies are then assigned to primary reviewers, secondary reviewers, and community member reviewers based on scientific content, proposed methods, planned engagement with human subjects and/or data about human subjects, and the required determinations necessary for IRB approval.
Full committee deliberations and determinations are guided by HRP-314: Criteria for Approval and Additional Considerations.
IRB Administration encourages reviewers to use the following outline during review to support comprehensive attention to human subjects considerations, compliance with applicable regulations, and consistency in full committee deliberations and actions.
Reviewer Outline
Background:
- Brief summary of the background, aims, number of arms, controls, randomization, etc.
- Investigator or research team conflicts of interest.
Risks & Benefits: (Criteria 1-3)
- Risks are minimized and reasonable in view of potential benefits.
- Research versus standard of care clearly outlined.
- Investigational drugs and/or devices appropriately described.
Subject Selection: (Criterion 4)
- Screening methods used (EMR) are described, and if necessary, a waiver of HIPAA for recruitment purposes is requested.
- Recruitment methods (who, where, when, by whom) clearly outlined.
- Recruitment materials are appropriate.
- Costs to subjects are outlined, with out of pocket expenses clearly delineated and justified.
- Compensation and/or reimbursement plan in place, with plan to prorate.
- Number of subjects reasonable/statistically justified.
Data Monitoring: (Criterion 5)
- Adequate provisions for monitoring data (DSMB).
- Plans for data and statistical analysis are defined and justified, including stopping rules and end points.
Privacy & Confidentiality: (Criteria 6 & 7)
- Provisions to protect the confidentiality and privacy of subjects.
- Adequate plans to store and code the data.
- If identifiers are used, or links to identifiers are necessary, identify how the information is protected.
Vulnerable Populations: (Criterion 8)
Targeting children, and adequate provisions to protect vulnerable subjects.
Targeting cognitively impaired adults, and adequate provisions to protect vulnerable subjects.
- HRP-417: Cognitively Impaired Adults
- Maximization of subject capacity for consent, if any
- Surrogate consent
Targeting pregnant women, and adequate provisions to protect vulnerable subjects.
Targeting neonates, and adequate provisions to protect vulnerable subjects.
Targeting prisoners, and adequate provisions to protect vulnerable subjects.
Consent Process: (Criterion 9)
- Process for obtaining consent is adequately described.
- Surrogate consent.
Consent Document: (Criterion 10)
- All elements of consent included.
- Readability for the subject group (including children).
- Separate assent form due to complexity of the study.
Expiration Dates
Initial and continuing review submissions are typically approved for a period of one year from the date of the committee meeting or administrative approval (in the case of approval contingent on required modifications). The committee may grant a shorter period of approval if they feel that it is warranted due to potential risks to subjects, but the approval period granted by the full committee cannot exceed one year.
IRB Fees
The Office of Research will charge Institutional Review Board (IRB) fees for new clinical research submissions that are partially or fully supported by industry sponsors, including chart review studies. More information available here .
Becoming an IRB Member
The IRB committees have members across all departments and many disciplines. Faculty and staff with various areas of expertise are always needed. A term of service usually begins with a one year probationary period and a two-year term thereafter. The contribution of time and expertise is invaluable to the university and its overall commitment to the protection of the rights and welfare of subjects who volunteer to participate in biomedical and behavioral research.
Biomedical/Clinical Committee A meets virtually on the first and third Wednesday of the month from 12:00-3:30.
Biomedical/Clinical Committee B meets virtually on the second and fourth Monday of the month from 12:00-3:30.
If you are interested in serving on these committees or would like more information on becoming a member, please contact the IRB.
Special Situations
Some types of research will require additional documentation or ancillary approvals prior to review. Please contact the IRB if your study involves any of these things:
- Wards of the court or the Division of Juvenile Justice
- Prisoners
- Funding from the National Institute of Justice (NIJ) , Department of Justice (DOJ) or Department of Defense (DOD)
- Exception from informed consent for emergency research
- Emergency or compassionate use of an unapproved drug or device
- Humanitarian use of an unapproved device