Reporting to the IRB – What You Need to Know
Investigators are required to report certain events that occur during the conduct of a study to the UC Davis IRB. These events are referred to as reportable events. The terms reportable event and reportable new information (RNI) are synonymous. For the remainder of this page, we will use the term reportable event.
If any member of a study team becomes aware of a reportable event, the event must be reported to the IRB within either five (5) or ten (10) working days depending on the nature of the event. On this page, you will find information about what events require prompt reporting to the UC Davis IRB.
In This Section
- What Events Must Be Reported to the IRB within 5 Working Days?
- What Events Must Be Reported to the IRB within 10 Working Days?
- What is a Minor Deviation?
- Reporting Requirements for Reliance Agreements
- How Do I Decide if an Event is Reportable?
Events that require prompt reporting may be found in Appendix A of the Investigator Manual. The timeline for reporting starts when a member of the study team, whether that is the Principal Investigator, Co-Principal Investigator, or other study personnel, becomes aware of the event.
If information contained within a report has previously been submitted to the IRB, do not submit a second time unless the updated report includes new information that would affect or revise the IRB’s previous determination.
What Events Must Be Reported to the IRB within 5 Working Days?
Information that indicates a new or increased risk, or a new safety issue
Events or new information that fall under this category include, but are not limited to, the following:
- New information (e.g., an interim analysis, safety monitoring report, publication, sponsor report, or investigator finding) indicating an increase in the frequency or magnitude of a previously known risk, or reveals a new risk.
- Failure to follow the protocol due to the action or inaction of anyone conducting protocol procedures (e.g., nursing staff, investigational pharmacy staff, and laboratory staff) that resulted in harm to subjects or others or that indicates subjects or others might be at increased risk.
- Subject complaint that indicates subjects or others might be at increased risk of harm or at risk of a new harm.
- An investigator’s brochure, package insert, or device labeling is revised to indicate an increase in the frequency or magnitude of a previously known risk, or describe a new risk.
- Withdrawal, restriction, or modification of a marketed approval of a drug, device, or biologic used in a protocol.
- Changes significantly affecting the conduct of the clinical trial or increasing the risk to participants.
Serious harm experienced by a subject or other individual, which in the opinion of the investigator and/or sponsor is unexpected and probably related (i.e., more than 50% likely) to the research procedures
IRB Tip: In the event of a disagreement, the sponsor’s assessment supersedes the Principal Investigator’s assessment.
What is a serious harm?
A harm is serious when it meets any of the following criteria:
- results in death,
- is life-threatening (places the subject at immediate risk of death from the event as it occurred),
- results in inpatient hospitalization or prolongation of existing hospitalization,
- results in a persistent or significant disability/incapacity,
- results in a congenital anomaly/birth defect,
- based upon appropriate medical/psychological judgment, may jeopardize the subject’s health and may require medical, counseling, or surgical intervention to prevent one of the other outcomes listed in this definition, or
- results in criminal or civil liability or damaging of the subject’s financial standing, employability, or reputation.
When is a serious harm unexpected?
A serious harm is unexpected when its specificity or severity are inconsistent with risk information previously reviewed and approved by the IRB in terms of nature, severity, frequency, and characteristics of the study population. Previously reviewed and approved risk information may be found in the following documents:
- Informed Consent Forms
- Investigator’s Brochures
- Package Inserts
- Instructions For Use
When is a serious harm probably related?
A serious harm is probably related to the research procedures if, in the opinion of the investigator and/or sponsor, the research procedures more likely than not caused the harm.
Anything less than 50% likely related is considered “possibly related” or unknown. Unless otherwise indicated by the sponsor, possibly related adverse events (including those that are unexpected and serious) do not need to be submitted to the IRB. However, should a follow-up report be received that changes the relatedness from possibly to related or probably related, you must submit a copy of the adverse event report within 5 working days.
Change to the protocol done without prior IRB review to eliminate an apparent immediate hazard to a subject
If it is possible to have the IRB review the change to the protocol prior to the change being made (i.e., a planned protocol deviation), a modification should be submitted instead of a reportable event. Instructions for submitting a planned protocol deviation may be found on the Planned Protocol Deviations webpage.
Unanticipated Adverse Device Effect (UADE)
An Unanticipated Adverse Device Effect (UADE) is any serious adverse effect on health or safety or any life-threatening problem or death caused by, or associated with, a device, if that effect, problem, or death, was not previously identified in a nature, severity, or degree of incidence in the investigational plan or application (including a supplementary plan or application), or any other unanticipated serious problem associated with a device that relates to the rights, safety, or welfare of subjects.
Non-Compliance
Non-compliance with the federal regulations governing human research or with the requirements or determinations of the IRB that pose a harm to the rights, safety, or welfare of the subject, or to the integrity of the data. Non-compliance is the result of the action or inaction of anyone conducting research procedures but not study subjects.
For more information about reporting non-compliance, please refer to the Corrective and Preventative Action Plans (CAPAs) webpage.
If non-compliance resulted in harm to subjects or others or if non-compliance indicates subjects or others might be at increased risk, then it must be reported to the UC Davis IRB within 5 working days.
Failure to follow the protocol due to the action or inaction of anyone conducting protocol procedures that results in harm to the integrity of the research data
Events in this category are oftentimes referred to as protocol deviations. Protocol deviations may be due to anyone conducting protocol procedures, including nursing staff, investigational pharmacy staff, and clinical laboratory staff.
However, this category of reportable event does not include protocol deviations due to action or inaction of a study subject. Such protocol deviations are considered minor deviations. Minor deviations are not reportable to the UC Davis IRB.
Note: If the protocol deviation resulted in harm to subjects or others or if the protocol deviation indicates subjects or others might be at increased risk, then it must be reported to the UC Davis IRB within 5 working days.
Subject complaint that cannot be resolved by the research team
If the subject’s complaint can be resolved by the research team, then it does not need to be reported to the UC Davis IRB.
Breach of confidentiality
A breach of confidentiality is any inappropriate disclosure of or access to confidential information.
If there is a breach of confidentiality involving Protected Health Information (PHI), report this breach to UC Davis Health Systems Compliance in addition to the UC Davis IRB.
Premature suspension or termination by the sponsor, investigator, or institution
This category includes the premature suspension or termination of specific treatment arms or sub-studies.
If premature suspension or termination indicates subjects or others might be at increased risk, then it must be reported to the UC Davis IRB within 5 working days.
Incarceration of a subject in a study not approved by the IRB to involve prisoners
You may confirm if your study is approved to enroll prisoners by reviewing the Initial Review Application submitted to the IRB.
Approvals and reports
Examples of events that fall under this category include the following:
- Ancillary approvals (e.g., Certificate of Confidentiality or Stem Cell Research Oversight Committee) that do not result in a protocol revision
- Data Safety Reports that include a recommendation to terminate or modify a study
- Written reports by study monitors and auditors that include reportable events that have not yet been reported
If a report indicates subjects or others might be at increased risk, then it must be reported to the UC Davis IRB within 5 working days.
Inquiry by a federal agency and any reports
Any inquiries or reports from a federal agency, such as the FDA Form 483, are included in this category.
This category is for any information or events that do not meet any of the above criteria. For example, if a sponsor requests for a specific event to be reviewed by the UC Davis IRB.
Other
This category is for any information or events that do not meet any of the above criteria. For example, if a sponsor requests for a specific event to be reviewed by the UC Davis IRB.
What is a Minor Deviation?
Minor deviations are protocol deviations that do NOT need to be reported to the UC Davis IRB because they do NOT pose a risk of harm to the subject’s rights, safety, or welfare, or to the integrity of the research data. Minor Deviations may result from the action or inaction of anyone conducting research procedures or the participant. Below is a decision tree to help you determine if a protocol deviation is a minor deviation:
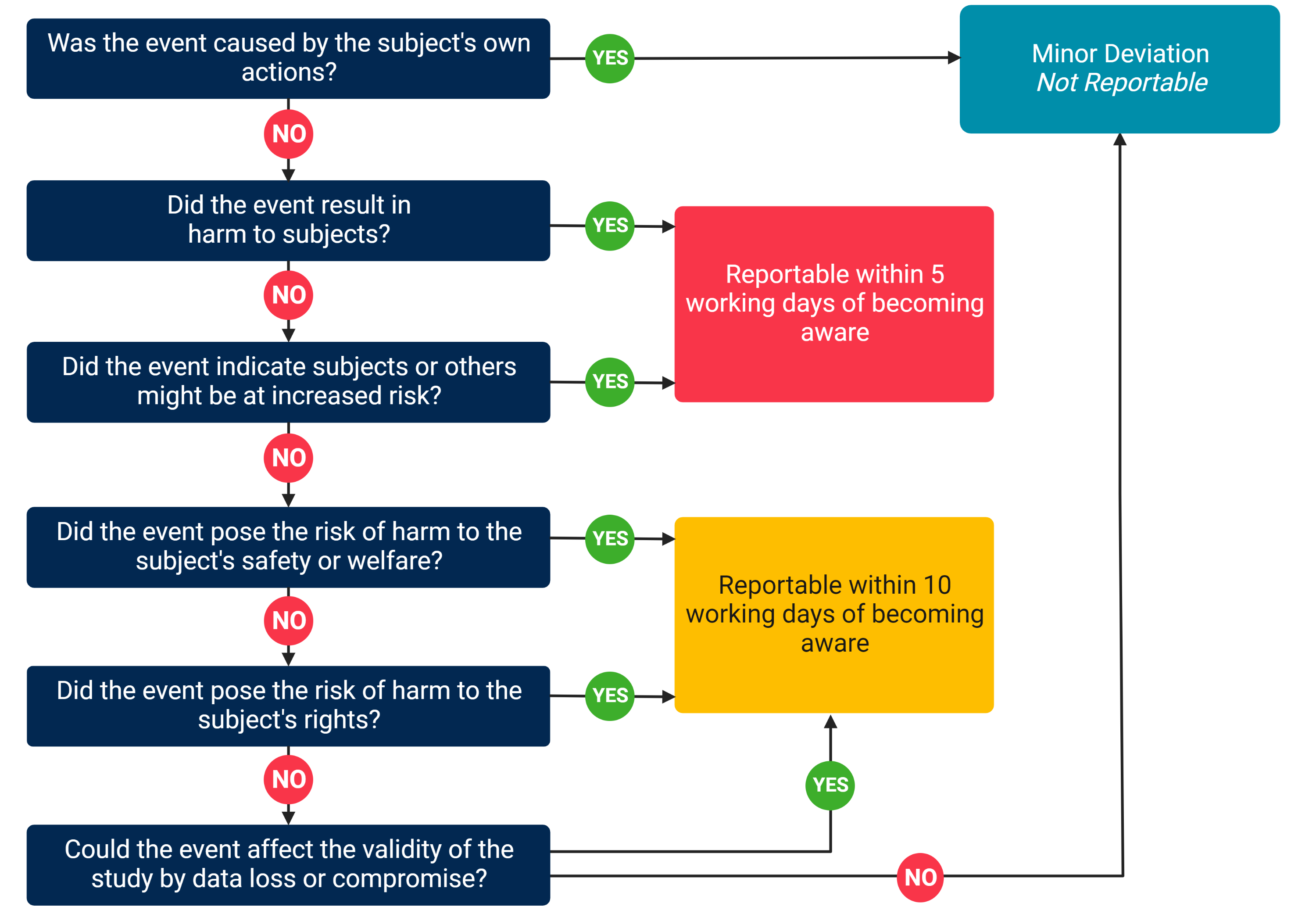
Some common examples of minor deviations to be aware of include:
- Missing or incorrect dates on signed informed consent forms
- Over-enrollment of subjects in minimal risk studies
- Continuing reviews submitted 15 days or less after the administrative due date
CITI training lapsed 30 days or less as long as no research procedures were conducted by the research staff with expired training that could pose a risk of harm to the subject’s rights, safety, or welfare, or to the integrity of the research data. Examples of such procedures include the following:
- Obtaining informed consent
- Collecting vital signs via direct contact
- Administering study drug
- Signing off on case report forms
If a sponsor requires that minor deviations are reported to the UC Davis IRB, they may be included in the Project Events Summary Table at the time of annual renewal. Regardless of sponsor requirements, we recommend maintaining a log of minor deviations in the research file.
Reporting Requirements for Reliance Agreements
Reporting requirements when UC Davis is Relying on an External IRB
When UC Davis is relying on an external IRB for review, reportable events must be reported to the reviewing IRB according to the reviewing IRB’s reporting requirements.
In the event the reviewing IRB makes any of the following determinations, please notify the UC Davis IRB:
- Suspension
- Serious Non-Compliance
- Continuing Non-Compliance
- Unanticipated Problems Involving Risk to Subjects or Others
Reporting requirements when UC Davis is the IRB of Record
When UC Davis is the reviewing IRB, reportable events must be submitted to the UC Davis IRB in accordance with the reporting requirements on this webpage. These reporting requirements apply to UC Davis researchers and all external relying sites.
How Do I Decide if an Event is Reportable?
The case studies provided below are designed to help you identify whether a specific event is reportable to the UC Davis IRB. These case studies do NOT represent an exhaustive list of reportable events. If you have any questions about whether an event is reportable to the UC Davis IRB, contact [email protected].
Protocol Deviations
The Principal Investigator of a study misreads the protocol and fails to order the appropriate dose of an investigational antibiotic. As a result, the subject’s infection worsens and she becomes septic.
Reportable within 5 Business Days
This event resulted in harm to the subject. As such, this event falls under Item 1b in Appendix A of the Investigator Manual:
Failure to follow the protocol due to the action or inaction of anyone conducting protocol procedures (e.g., nursing staff, IDS pharmacy staff, and laboratory staff) that resulted in harm to subjects or others or that indicates subjects or others might be at increased risk.
A researcher fails to administer an electrocardiogram (EKG) at a study visit as required by the protocol. Per the protocol, the EKG at this visit is a safety-related procedure. In the Principal Investigator’s opinion, the subject did not experience any harm due to the missed EKG.
Reportable within 10 Business Days
Although the missed EKG did not result in harm to the subject, since the purpose of the EKG is related to subject safety, failure to administer the EKG at this visit poses a harm to the subject’s safety. As such, this event falls under Item 6 in Appendix A of the Investigator Manual:
Failure to follow the protocol due to the action or inaction of anyone conducting protocol procedures (e.g., nursing staff, IDS pharmacy staff, and laboratory staff) that poses a harm to the rights, safety, or welfare of the subject, or to the integrity of the data.
A survey is administered to a subject outside of the window described in the protocol. In the Principal Investigator’s opinion, this event did not pose a harm to the rights, safety, or welfare of the subject or the integrity of the data.
Not Reportable (Minor Deviation)
This event is not reportable because it constitutes a minor deviation as described in SOP HRP-001 Definitions:
A change to, or non-compliance with, the research protocol that does not pose a risk of harm to the subject’s rights, safety or welfare, or to the integrity of the research data. Minor Deviations may result from the action or inaction of the participant, researcher, or research staff.
A subject oversleeps for a scheduled study visit. At this study visit, the subject was supposed to have blood drawn for several safety-related labs.
Not Reportable (Minor Deviation)
Although safety-related labs were to be drawn at this visit, this event is not reportable because it constitutes a minor deviation as defined in SOP HRP-001 Definitions since it resulted from inaction of the subject instead of the research team:
A change to, or non-compliance with, the research protocol that does not pose a risk of harm to the subject’s rights, safety or welfare, or to the integrity of the research data. Minor Deviations may result from the action or inaction of the participant, researcher, or research staff.
Informed Consent Forms (ICFs)
A researcher conducting a non-exempt study accidentally uses a version of the most recently approved ICF without a stamp to obtain consent from a participant.
Not Reportable
As long as the most recently approved version of the ICF was used, the use of an ICF without a stamp is not reportable to the UC Davis IRB. Please note, consent documents for exempt studies are not stamped.
For more information about documentation of informed consent, please refer to SOP HRP-091 Written Documentation of Informed Consent.
A researcher does not use the most recently approved version of the ICF to obtain consent from a participant. The most recently approved version of the ICF includes relevant information that would affect the subject’s willingness to participate in the study (e.g., study procedures, risks, alternatives). This information was not included in the version of the ICF used to obtain consent from the subject.
Reportable within 10 Business Days
Potential subjects have the right to be informed of all information that would affect their willingness to participate in a study. As such, this event represents a harm to the subject’s rights and is considered non-compliance. Non-compliance is Item 5 in Appendix A of the Investigator Manual:
Non-compliance with the federal regulations governing human research or requirements or determinations of the IRB that pose a harm to the rights, safety, or welfare of the subject, or to the integrity of the data.
A researcher does not use the most recently approved version of the ICF to obtain consent from a participant. The only difference between the outdated ICF and the most recently approved ICF was the correction of some typographical errors that did not alter the meaning of the form.
Not Reportable
As opposed to the previous case, this event does not represent non-compliance because no harm was posed to the rights, safety, or welfare of the subject, or to the integrity of the data.
Investigator’s Brochure Updates
A researcher receives a new version of the Investigator‘s Brochure from a sponsor. The risks in the Investigator’s Brochure were updated to include the risk of cardiac arrhythmia.
Reportable within 5 Business Days
This Investigator’s Brochure update represents information that indicates there is a new risk, cardiac arrhythmia. As such, this information falls under Item 1a in Appendix A of the Investigator Manual:
New information (e.g., an interim analysis, safety monitoring report, publication, sponsor report, or investigator finding) indicating an increase in the frequency or magnitude of a previously known risk, or reveals a new risk.
A researcher receives a new version of the Investigator’s Brochure from a sponsor for a study. Both the sponsor and Principal Investigator confirm that there were no new risks or increases in the frequency or magnitude of a previously known risk.
Not Reportable
Since both the sponsor and Principal Investigator agree that this Investigator’s Brochure update does not include information that indicates that there are new risks or increases in the frequency or magnitude of a previously known risk, it is not reportable to the UC Davis IRB.
A researcher receives a new version of the Investigator‘s Brochure from a sponsor for a study that is closed to accrual. Subjects at UC Davis are no longer receiving the investigational drug. The risks in the Investigator’s Brochure were updated to include the risk of psychosis. The sponsor and Principal Investigator agree that the risk of psychosis is a long-term risk.
Reportable within 5 Business Days
Given that psychosis represents a long-term risk, this Investigator’s Brochure update represents information that indicates a new risk for these subjects. As such, this information falls under Item 1a in Appendix A of the Investigator Manual:
New information (e.g., an interim analysis, safety monitoring report, publication, sponsor report, or investigator finding) indicating an increase in the frequency or magnitude of a previously known risk, or reveals a new risk.
A researcher receives a new version of the Investigator‘s Brochure from a sponsor for a study that is closed to accrual. Subjects at UC Davis are no longer receiving the investigational drug. The risks in the Investigator’s Brochure were updated to include the risk of an immune reaction immediately after administration of the investigational drug. The sponsor and Principal Investigator agree that the risk of an immune reaction is not a long-term risk.
Not Reportable
As opposed to the previous case, an immune reaction does not represent a long-term risk. This Investigator’s Brochure update does not indicate there is a new risk for these subjects.
Serious Adverse Events (SAEs)
A researcher receives a report of a serious adverse event from a sponsor. The report states that the event was unexpected. However, it was deemed not related to the research procedures.
Not Reportable
Adverse events are reportable to the UC Davis IRB only if they are serious, unexpected, and at least probably related (>50% likely) to study procedures.
A researcher receives a report of a serious adverse event from a sponsor. The report states that the event was unexpected and probably related to the research procedures.
Reportable within 5 Business Days
Adverse events are reportable to the UC Davis IRB only if they meet all of the following criteria:
- Serious
- Unexpected
- Related or probably related (>50% likely) to the research procedures
As such, this information falls under Item 2 in Appendix A of the Investigator Manual:
Serious harm experienced by a subject or other individual, which in the opinion of the investigator is unexpected and probably related (i.e., more than 50% likely) to the research procedures.
Data Safety Monitoring Board (DSMB) or Data Safety Monitoring Committee (DSMC) Reports
A researcher receives a DSMB/C report, which states that the study may continue as planned. There were no changes to risk information.
Not Reportable
Since the DSMB/C report does not include information that indicates that there are new risks or increases in the frequency or magnitude of a previously known risk, it is not reportable to the UC Davis IRB.
A researcher receives a DSMB/C report. One of the study arms is being discontinued because the DSMB/C’s analysis revealed an increased frequency of strokes in subjects randomized to this arm.
Reportable within 5 Business Days
Given that subjects in this arm are experiencing an increased frequency of stroke, this DSMB/C report represents information that indicates an increase in the frequency of a known risk. As such, this information falls under Item 1a in Appendix A of the Investigator Manual:
New information (e.g., an interim analysis, safety monitoring report, publication, sponsor report, or investigator finding) indicating an increase in the frequency or magnitude of a previously known risk, or reveals a new risk.
A researcher receives a DSMB/C report. The study is being terminated before all subjects were enrolled because the lead site has run out of funding to support the study.
Reportable within 10 Business Days
As opposed to the previous case, this DSMB/C report does not represent information that indicates an increase in the frequency of a known risk because the study is being terminated for a non-safety reason. As such, this information falls under Item 9 in Appendix A of the Investigator Manual:
Premature suspension or termination by the sponsor, investigator, or institution.
Miscellaneous
A laptop with identifiable research data is stolen from a researcher’s car.
Reportable within 10 Business Days
This event represents a breach of confidentiality because the subjects’ identifiable data is now accessible to those not involved in the research. As such, this event falls under Item 7 in Appendix A of the Investigator Manual:
Breach of confidentiality (inappropriate disclosure of or access to confidential information).
A subject complains to a researcher that he never received a gift card for his participation in a study as described in the informed consent form. The researcher resolves the compensation issue by providing this subject with a gift card.
Not Reportable
Since this complaint was resolved by the research team, it is not reportable to the UC Davis IRB.
A subject complains to a researcher that he never received a gift card for his participation in a study as described in the informed consent form. The researcher is unable to resolve the compensation issue because the study has now exhausted all of its funding.
Reportable within 10 Business Days
As opposed to the previous case, this complaint could not be resolved by the research team. As such, this event falls under Item 8 in Appendix A of the Investigator Manual:
Complaint of a subject that cannot be resolved by the research team.